Research Methodology
|
Our research is rooted in comparative genomics: we employ genetic biodiversity as the key resource to untangle evolutionary processes, and understand the underlying genetic mechanisms and molecular functions. This approach is particularly relevant nowadays, as new species are sequenced at unprecedented pace (see Figure 1). Our research encompasses diverse evolutionary timescales, from the deep roots of life to recent population histories.
We typically use bioinformatics as first method of discovery, and then we bring hypotheses and resources to the lab. Our bioinformatic expertise includes a variety of techniques from evolutionary genomics such as gene prediction, phylogenetics, sequence analysis, transcriptomics, and quantitative data analysis. On the experimental side, we employ cell cultures from mammals and insects, and also we perform RNAseq and ribosome profiling. |
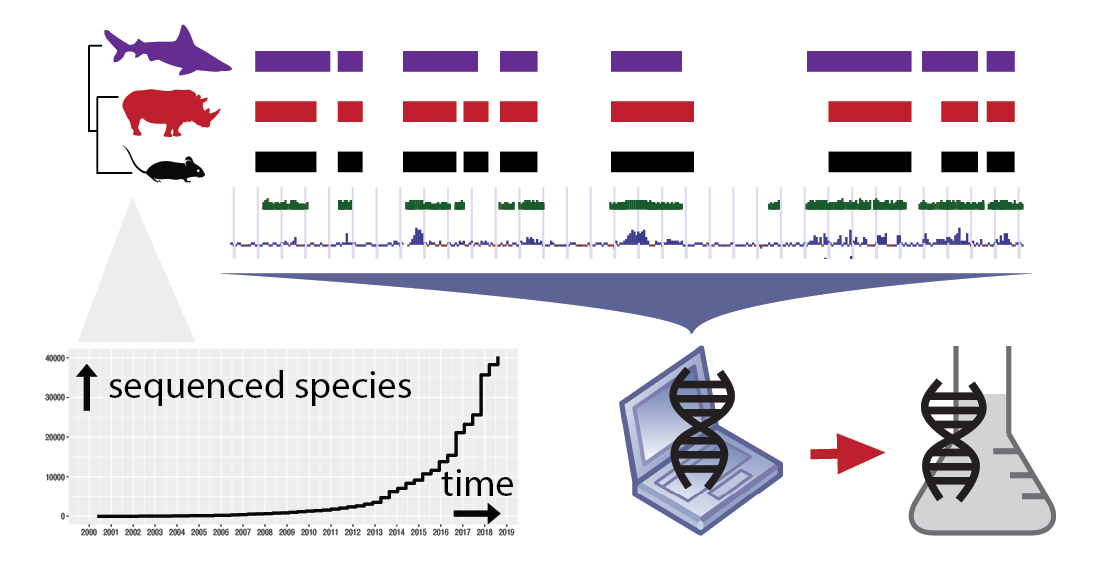
Figure 1.
|
Research Themes
Introduction: What is translational recoding?
|
Translation is a fundamental process ubiquitous to life. The genetic code defines how mRNA sequences are translated into proteins, with unequivocal correspondences between codons (nucleotide triplets) and 20 proteinogenic amino acids. Certain genes, however, are translated with systematic deviations to the genetic code used for the rest of the genome. Such programmed exceptions to protein synthesis are known as recoding.
Recoding is widespread throughout all domains of life, and many forms are known. Most notably, stop codon readthrough is a form of recoding wherein a stop supports amino acid insertion instead of translation termination (see Figure 2). "Canonical readthrough" occurs through stop codon recognition by a near-cognate tRNA, resulting in insertion of a standard amino acid. Moreover, two non-canonical amino acids exist, selenocysteine (Sec) and pyrrolysine (Pyl), inserted by peculiar forms of readthrough. These involve specialized genetic machinery, including a dedicated tRNA with anticodon complementary to a stop. Pyl usage is limited to a small number of prokaryotes, while Sec is much more common. |
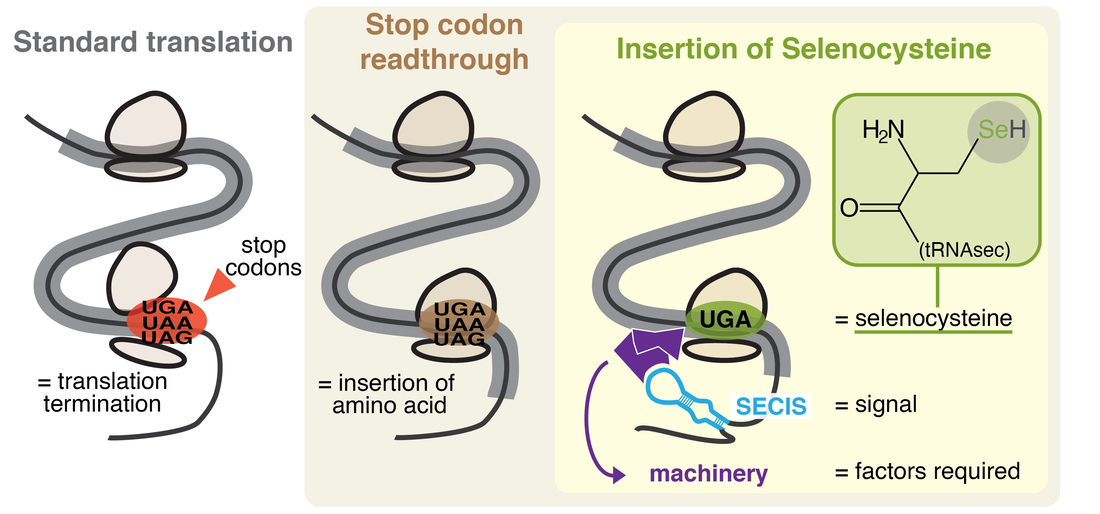
Figure 2.
|
Selenocysteine: the 21st amino acid
|
Selenocysteine (Sec) is a non-standard amino acid encoded by the stop codon UGA. Sec-containing proteins are called selenoproteins. UGA is recoded for Sec insertion by a specific readthrough mechanism requiring a cis-signal present in selenoprotein transcripts (the SECIS element). In selenoproteins, Sec is generally found in the catalytic site, where it provides catalytic advantages compared to standard amino acids. Selenoproteins have various essential roles for human health, and most of them are involved in redox homeostasis.
|

Figure 3.
|
Abundant stop codon readthrough in insects
|
Genes with "canonical" stop codon readthrough (i.e., other than Sec) are known in the most diverse organisms. Yet, insects constitute a truly remarkable case: recent research has uncovered hundreds of genes with conserved readthrough in insect genomes (e.g. ~900 in Drosophila). Very little is known about the mechanism and function of abundant readthrough in this lineage. We hypothesize that many molecular novelties lurk undiscovered in this unexplored diversity. Therefore, we are working to investigate this process at large by combining bioinformatics and experiments (see Figure 4):
|
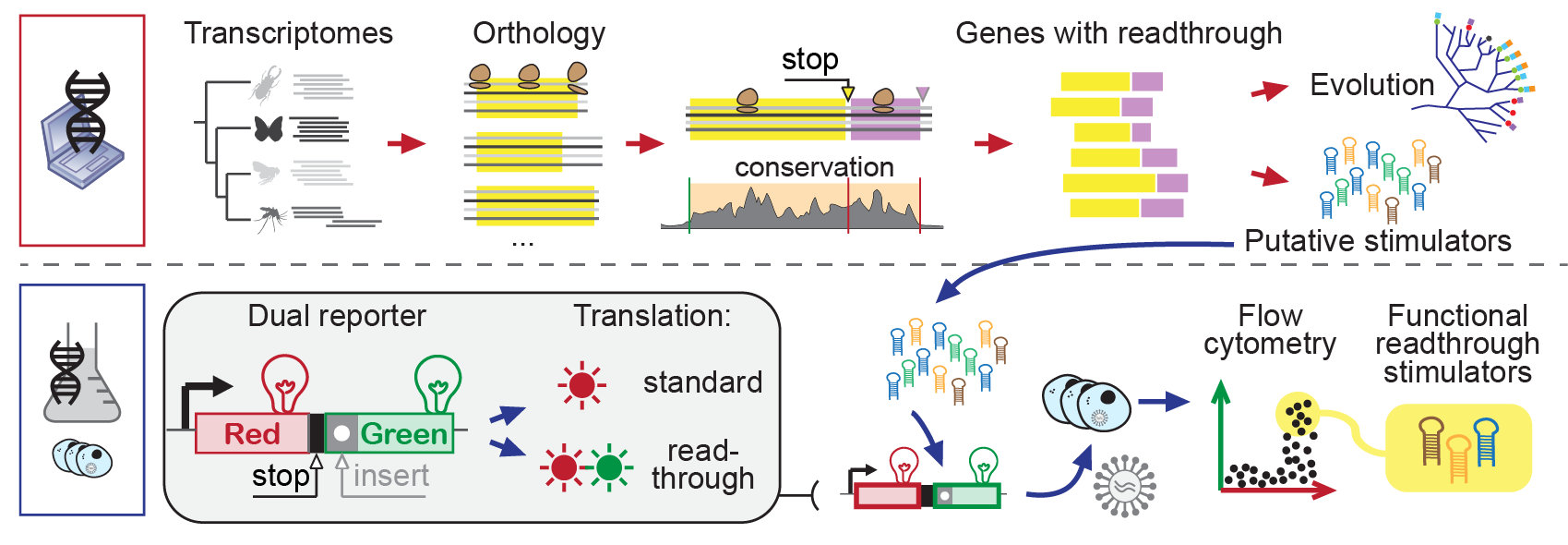
Figure 4.
|
Bioinformatics tools for genomics
|
Our lab develops bioinformatics tools for genomics. Some are focused on our research topics, while others have more general purposes. Visit the software page for more information. Most notably:
|
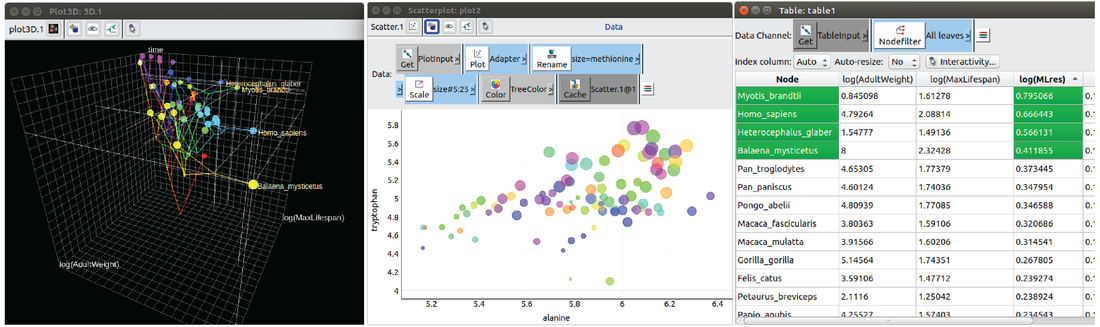
Figure 5.
|